
Analytical Method Development and Validation
Table of Contents
Analytical method development, validation, and transfer are key elements of any pharmaceutical development program. This technical brief will focus on development and validation activities applicable to drug products. Often considered routine, the benefit that well-developed analytical methods can contribute to the overall developmental time and cost efficiency of a program is undervalued.
Method-related activities are interrelated. They are also iterative particularly during early drug development phases. Parts of each process may occur concurrently or be refined at various phases of drug development. Changes to one method during drug development may require modifications to a separate existing analytical method. These modifications in turn may require additional validation or transfer activities, as shown in Figure 1.
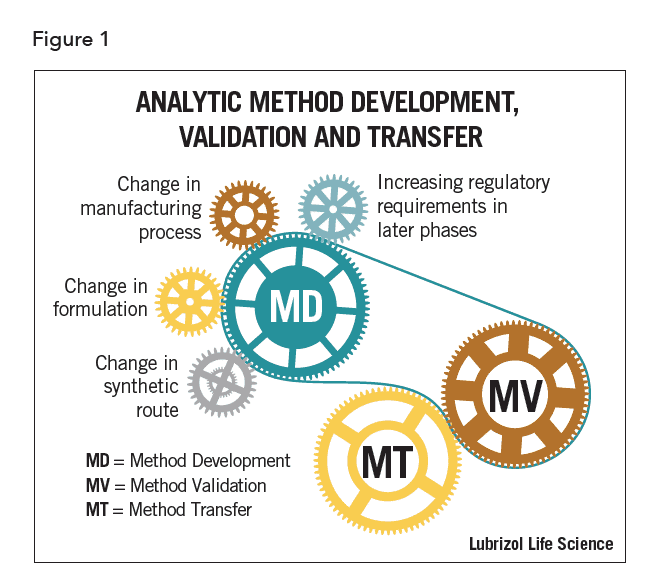
Effective method development ensures that laboratory resources are optimized, while methods meet the objectives required at each stage of drug development. Method validation, as required by regulatory agencies at certain stages of the drug approval process, is defined as the “process of demonstrating that analytical procedures are suitable for their intended use”1. Method transfer is the formal process of assessing the suitability of methods in another laboratory. Each of these processes contributes to continual improvement of methods and results in more efficient drug development.
Analytical methods are intended to establish the identity, purity, physical characteristics and potency of drugs. Methods are developed to support drug testing against specifications during manufacturing and quality release operations, as well as during long-term stability studies. Methods may also support safety and characterization studies or evaluations of drug performance. According to the International Conference on Harmonization (ICH), the most common types of analytical procedures are: (i) identification tests, (ii) quantitative tests of the active moiety in samples of API or drug product or other select- ed component(s) in the drug product, (iii) quantitative tests for impurity content, and (iv) limits tests for the control of impurities2.
Method development (Figure 2) is a continuous process that progresses in parallel with the evolution of the drug product. The notion of phase-appropriate method development is a critical one if time, cost and efficiency are concerns. The goal and purpose of the method should reflect the phase of drug development. During early drug development the methods may focus on API behavior. They should be suitable to support pre-clinical safety evaluations, pre-formulation studies, and prototype product stability studies. As drug development progresses, the analytical methods are refined and expanded, based on increased API and drug product knowledge. The methods should be robust and uncomplicated, while still meeting the appropriate regulatory guidelines.
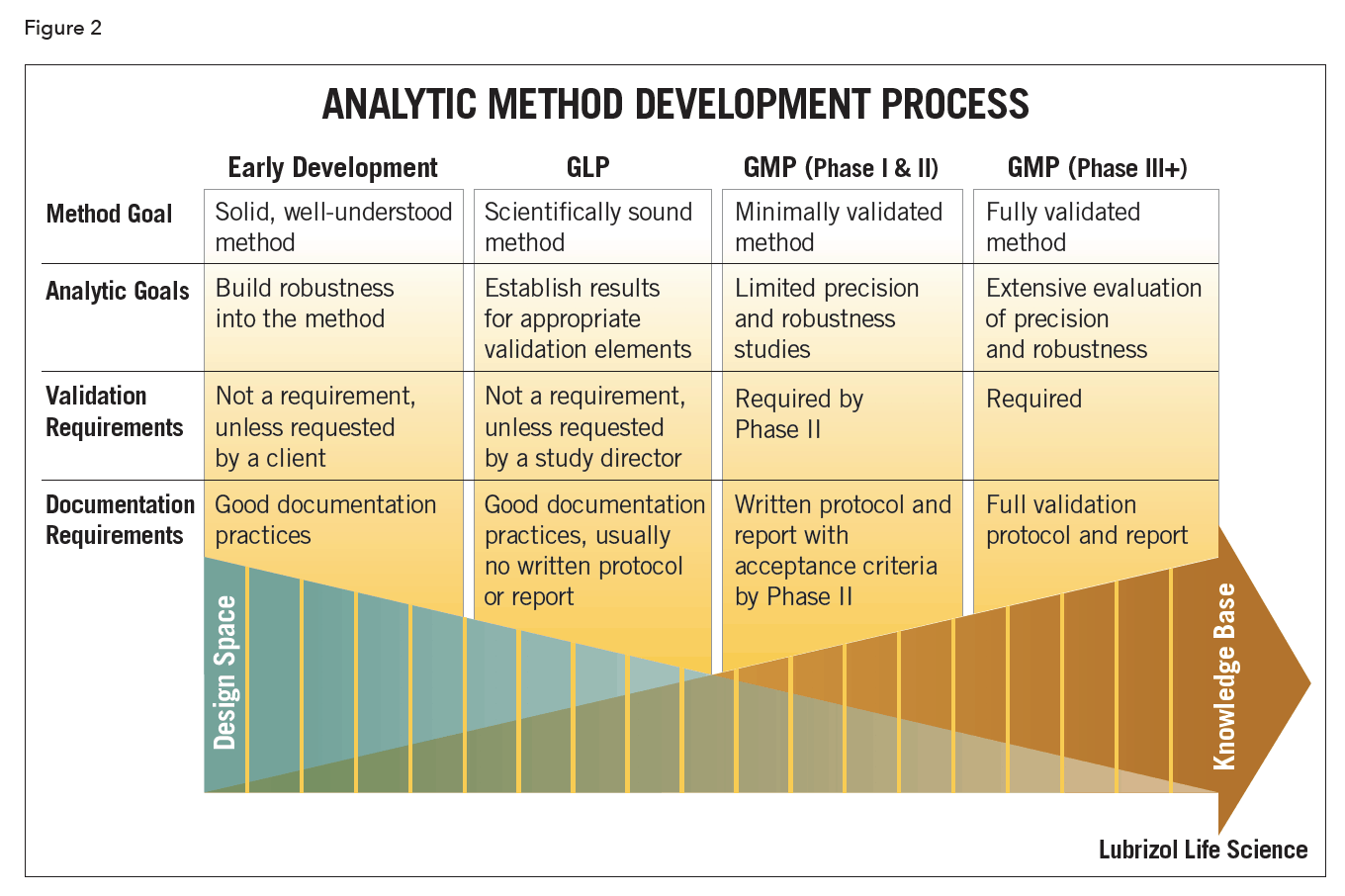
Scouting experiments are frequently performed during method development to establish the performance limits of the method prior to formal validation experiments. These may include forced degradation studies, which are an integral part of development of a stability-indicating method. API is typically subjected to degradation by acid, base, oxidation, heat, and light. This allows for a determination of the capability of the method to separate and quantify degradation products, while providing insight into the main mechanisms of degradation. Once a stability-indicating method is in place, the formulated drug product can then be subjected to heat and light to evaluate potential degradation of the API in the presence of formulation excipients.
Additional experiments help to define the system suitability criteria that will be applied to future analytical sample sets. System suitability tests are a set of routine checks to assess the functionalities of the instrument, software, reagents, and analysts as a system3. Final method system suitability parameters may be determined from evaluations of method robustness performed under statistical design of experiments. The goal is to identify the critical parameters and establish acceptance criteria for method system suitability.
Elements of Validation
The validation of an analytical method demonstrates the scientific soundness of the measurement or characterization and is required throughout the regulatory submission process. The validation practice demonstrates that an analytical method measures the correct substance, in the correct amount, and in the appropriate range for the intended samples. It allows the analyst to understand the behavior of the method and to establish the performance limits of the method. Resources for information and approaches to method validation are listed in the endnotes
In order to perform method validation, the laboratory should be following a written standard operating procedure (SOP) that describes the process of conducting method validation. The laboratory should be using qualified and calibrated instrumentation with a corresponding operating SOP. There should be a well-developed and documented test method in place and an approved protocol should be in place prior to the execution of any validation experiments. The protocol is a plan that describes which method performance parameters will be tested, how the parameters will be assessed, and the acceptance criteria that will be applied. Finally, samples of API or drug product, placebos, and reference standards are needed to perform the validation experiments.
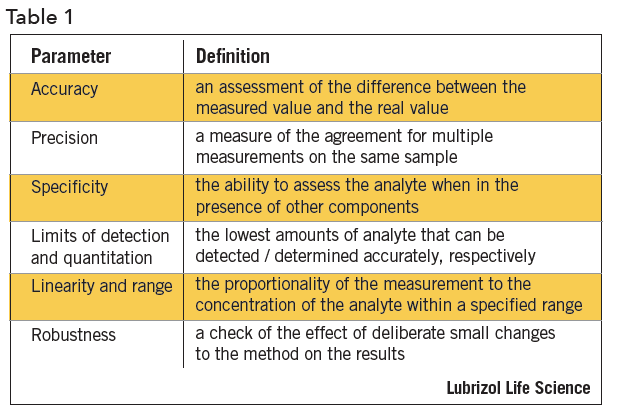
The method performance parameters that are applicable to most methods are shown in Table 14.
 Approaches to Validation Experiments
Approaches to Validation Experiments
 Approaches to Validation Experiments
Approaches to Validation ExperimentsAccuracy is established by quantitation of the sample against a reference standard for API, or spiking placebo with API for a drug product. It can also be determined by comparison of results from alternate measurement techniques.
Precision is determined by multiple measurements on an authentic, homogeneous set of samples. Samples may be analyzed on different days, by different analysts, on different instruments, or in different laboratories. There are three levels of precision validation evaluations – repeatability, intermediate precision, and reproducibility. Repeatability is a measure of precision under the same conditions over a short period of time. Intermediate precision is a measure of precision within the same laboratory by different operators, using different instruments, and making measurements on different days. Reproducibility assesses precision between two or more laboratories.
Specificity can be established by several approaches, depending on the intended purpose of the method. The ability of the method to assess the analyte of interest in a drug product is determined by a check for interference by a placebo. Specificity can be assessed by measurement of the API in samples that are spiked with impurities or degradants. If API-related compounds are not available, drug can be stressed or force-degraded to produce degradation products. In chromatographic separations, apparent separation of degradants may be confirmed by peak purity determinations, photodiode array, mass purity determinations, mass spectroscopy (MS), or by confirming separation efficiency using alternate column chemistry. During forced degradation experiments, degradation is targeted at 5 to 20% degradation of the API to avoid concerns about secondary degradation.
The limit of detection and limit of quantitation are based on measurement signal-to-noise ratios of 3 and 10, respectively. Standards or samples at concentrations near the expected limits are measured. Signal-to-noise can be generated by software, manually measured, estimated from standard deviation calculations, or empirically determined
Linearity is established by measuring response at various concentrations by a regression plot, typically by method of least squares. The response may require mathematical manipulation prior to linearity assessments. A visual inspection of the linearity plot is the best tool for examining proportionality of the response. The range is established by the required limits of the method and the point at which linearity is compromised.
Robustness is typically assessed by the effect of small deliberate changes to chromatographic methods on system suitability parameters such as peak retention, resolution, and efficiency. Experimental factors that are typically varied during method robustness evaluations include: (i) age of standards and sample preparations, (ii) sample extraction time, (iii) variations to pH of mobile phase, (iv) variation in mobile phase composition, (v) analysis temperature, (vi) flow rate, (vii) column lot and/or manufacturer, and (viii) type and use of filter against centrifugation. Robustness experiments are an ideal opportunity to utilize statistical design of experiments providing data-driven method control.
The ICH guidance on validation separates types of methods according to the purpose of the method and lists which evaluations are appropriate for each type2.
The ICH guidance also suggests detailed validation schemes relative to the intended purpose of the methods. It lists recommended data to report for each validation parameter. Acceptance criteria for validation elements must be based on the historical performance of the method, the product specifications, and must be appropriate for the phase of drug development.
Timing of Validation
As previously mentioned, the path to validation forms a continuum. It begins in the early phases of drug development as a set of informal experiments that establish the soundness of the method for its intended purpose. It is expanded in intensity and extent throughout the regulatory submission process into a fully-documented report that is required by NDA submission at Phase III and in support of commercial production. It is repeated whenever there is a significant change in instrumentation, method, specifications, and process, if applicable.
Conclusion
Analytical method development and validation are continuous and interconnected activities conducted throughout the drug development process. The practice of validation verifies that a given method measures a parameter as intended and establishes the performance limits of the measurement. Although apparently contradictory, validated methods produce results within known uncertainties. These results are crucial to continuing drug development, as they define the emerging knowledge base supporting the product.
The time and effort that are put into developing scientifically-sound, robust, and transferrable analytical methods should be aligned with the drug development stage. The resources that are expended on method validation must be constantly balanced with regulatory requirements and the probability for product commercialization.
References
- FDA Guidance for Industry – Analytical Procedures and Method Validation, Chemistry, Manufacturing, and Controls Documentation, Center for Drug Evaluation and Re- search (CDER) and Center for Biologics Evaluation and Research (CBER), August 2000.
- International Conference on Harmonization Quality Guidelines Q2(R1), Validation of Analytical Procedures, Text and Methodology, Parent guideline dated 27 Oct 1994, Complementary guideline on methodology dated 6 Nov 1996, incorporated November 2005
- USP 31 (2009): General Tests, Chapter 621 – Chromatography System Suitability, United States Pharmacopeial Convention (USP), Rockville, MD.
- Chan, C. C. et. al. (ed.), (2004), Analytical Method Validation and Instrument Performance Verification, Hoboken, NJ: John Wiley & Sons (Wiley Interscience).