
Dissolution Testing for OSD
Table of Contents
Dissolution testing is a requirement for all solid oral dosage forms and is used in all phases of development for product release and stability testing1. It is a key analytical test used for detecting physical changes in an active pharmaceutical ingredient (API) and the formulated product.
At early stages of development in vitro dissolution testing guides the optimization of drug release from formulations. Over the past 50 years dissolution testing has also been employed as a quality control (QC) procedure, in R&D to detect the influence of critical manufacturing variables, and in comparative studies for in vitro–in vivo correlation (IVIVC)2.
The FDA guidance on dissolution testing for immediate release solid oral dosage forms includes the use of the Biopharmaceutics Classification System (BCS) for biorelevant dissolution tests, which is based upon API solubility and permeability1,3. According to the BCS guidelines, in vitro dissolution testing may be a useful tool to forecast the in vivo performance of drug products and potentially reduce the number of bioavailability/bioequivalence studies required. The FDA guidance on scale-up and post- approval changes (SUPAC) for immediate release oral dosage forms recommends the use of in vitro dissolution to justify post-approval changes4.
Despite being deeply ingrained in the pharmaceutical and biotechnology industry, the basics of the dissolution test are often misunderstood. The test must be reproducible and highlight or discriminate significant changes in product performance.
The specific dissolution technique employed is determined by the dosage form characteristics and the intended route of administration. For solid dosage forms, industry standard dissolution testing methodologies are the United States Pharmacopoeia (USP) Apparatus 1 (basket) and the USP Apparatus 2 (paddle) (see Figure 1). Immediate-release, modified- release, and extended release tablets are usually tested in classical dissolution baths with USP 2 paddles. Floating capsules and tablets generally use USP 1 baskets. Other dissolution techniques and equipment include USP 3 (reciprocating cylinders), USP 4 (flow-through-cell), USP 5 (paddle-over-disk), USP 6 (cylinder) and USP 7 (reciprocating holders)5.
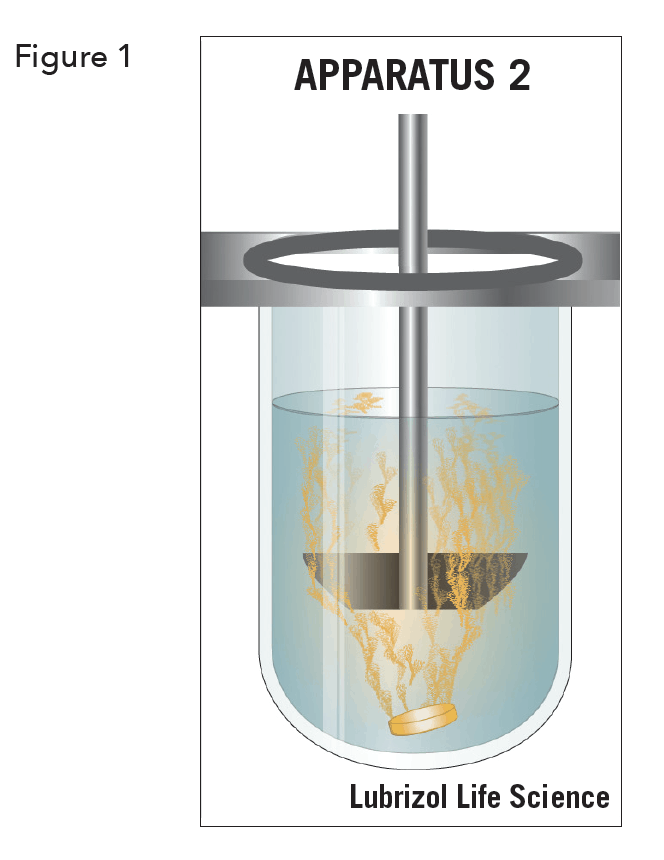 The development of a dissolution procedure involves selecting the dissolution media, apparatus type, and hydrodynamics (agitation rate) appropriate for the product. This overview article will focus on the most commonplace (USP 1 and 2) dissolution apparatuses and present an overview of typical method parameters to be considered during dissolution development.
The development of a dissolution procedure involves selecting the dissolution media, apparatus type, and hydrodynamics (agitation rate) appropriate for the product. This overview article will focus on the most commonplace (USP 1 and 2) dissolution apparatuses and present an overview of typical method parameters to be considered during dissolution development.
Dissolution
For most dosage forms to be efficacious, the API(s) must be introduced into the systemic circulation so that it can be transported to its site of activity. This process contributes to the bioavailability of the drug substance and involves two steps: dissolution and absorption (or permeability). Understanding the multi-step dissolution process is essential to proper in vitro method development. Dissolution is the process of extracting the API out of the dosage form solid-state matrix into solution within the gastrointestinal tract. Absorption is the process of transporting the drug substance from the gastrointestinal lumen into the systemic circulation.
Dissolution testing is an in vitro method that characterizes how an API is extracted out of a solid dosage form. It can indicate the efficiency of in vivo dissolution but does not provide any information on drug substance absorption. Pharmacokinetic data supplements and provides additional information regarding API absorption rate.
 Selection of the appropriate in vitro conditions (media and hydrodynamics) that simulate in vivo conditions can lead to the generation of successful IVIVC or, at the very least, in vitro–in vivo relations (IVIVR)3. Conditions that are optimal for QC purposes may not be applicable for establishing IVIVC, so it may be necessary to use two dissolution tests to meet different objectives, such as development needs or regulatory demands.
Selection of the appropriate in vitro conditions (media and hydrodynamics) that simulate in vivo conditions can lead to the generation of successful IVIVC or, at the very least, in vitro–in vivo relations (IVIVR)3. Conditions that are optimal for QC purposes may not be applicable for establishing IVIVC, so it may be necessary to use two dissolution tests to meet different objectives, such as development needs or regulatory demands.
Dissolution Method Parameters
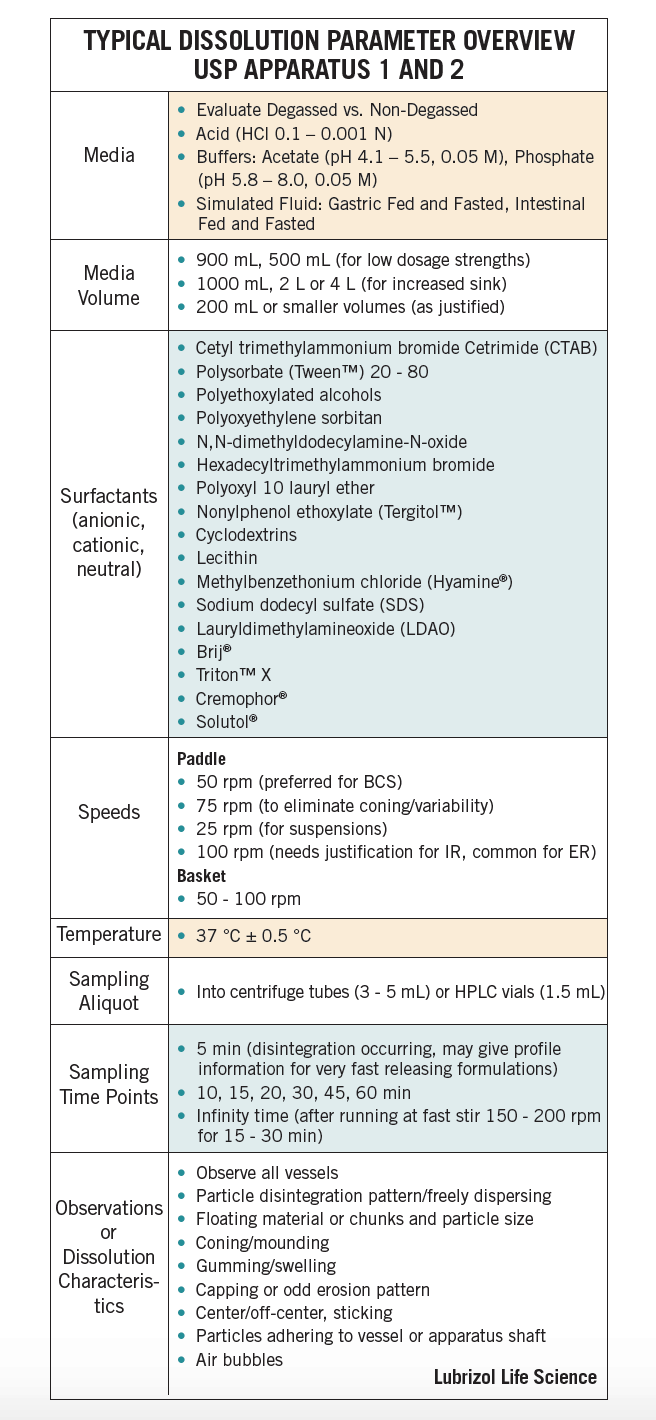
A logical, systematic approach taking into consideration both scientific and regulatory principles should be followed when developing a dissolution method. Table 1 lists common parameters and conditions that are evaluated during method development6.
A robust dissolution method must be free of artifacts, yield low-to-moderate variability, have good profile shape, and must be challenged to pick up critical quality attributes. Once the medium and apparatus are selected the method should be further optimized for parameters such as agitation rate, ionic strength, and surfactant concentration, if applicable. The final method should discriminate between formulations yet possess sufficient reproducibility and robustness. In terms of statistics, a relative standard deviation of <20% at early time points and <10% at later time points is common.
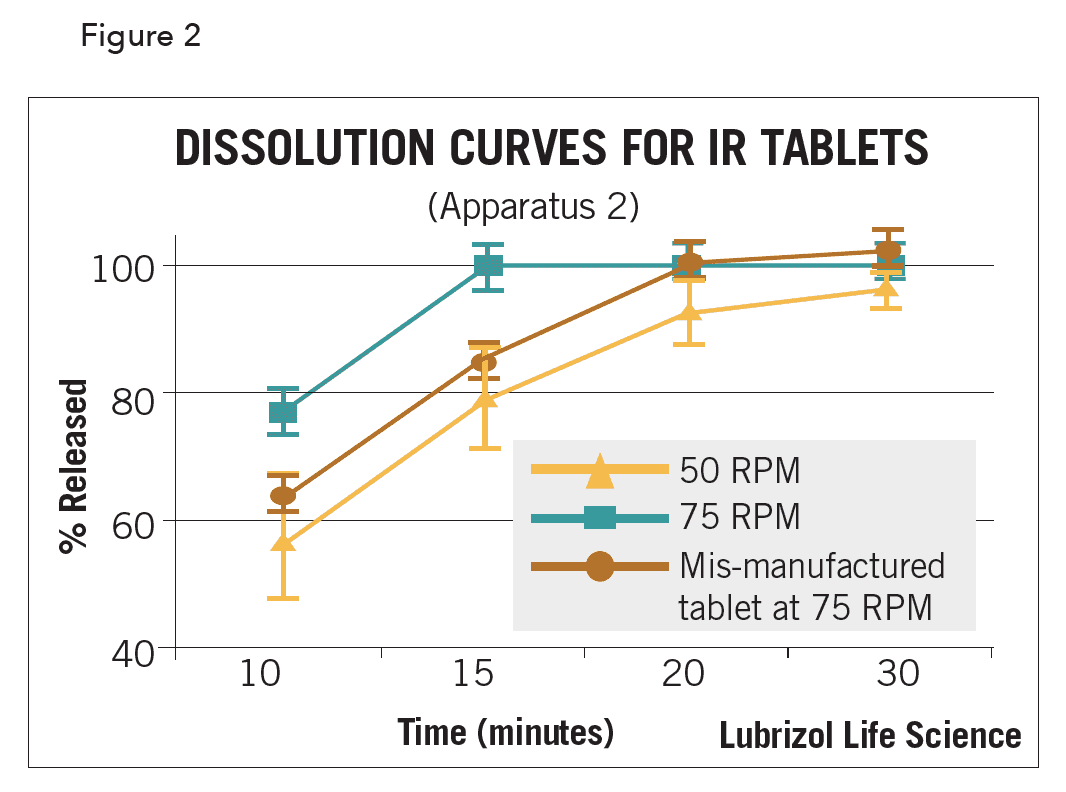
Typically, the percent dissolved API (up to 100%) vs. time is plotted. Dissolution profiles of dosage forms with known formulation, manufacturing, or bioavailability differences can aid in identifying a discriminatory set of media/hydrodynamic conditions. Figure 2 illustrates a dissolution plot at 50 rpm (tablet with increased release variability due to a method artifact known as coning), 75 rpm (tablet showing proper release), and a mis-manufactured tablet at 75 rpm (showing slower and improper/incomplete release).
API
Review of API properties (BCS-classification, pKa, stability, solubility as a function of pH/surfactant concentration, particle size, and polymorphisms) that are likely to affect the in vitro dissolution behavior should be evaluated as part of method development.
 Dosage Form
Dosage Form
The key properties of the dosage unit, including dosage form type (tablet, capsule), expected number of potencies, and desired release mechanism plus specific formulation information such as excipients, lubricants, disintegrants, moisture content, surface coating, and known stability issues (cross-linking, friability) are all important factors to consider. Manufacturing variables such as lubrication blend time, compression force, excipient/API addition order, drying parameters, and coating parameters are also critical to understanding API release differences between formulations.
Media
The first step is to screen formulations with aqueous-based media in the range of pH 1.2 to 6.8 at the USP recommended ionic strength.5 For APIs that exhibit low solubilities in aqueous media throughout the pH range, the addition of surfactants is recommended. A medium resulting in a gradual increase of released drug up to 100% is preferred because it is more likely to detect differences in formulation or processing parameters.
Visual Observations
It is imperative to visually observe the behavior of the dosage form throughout the dissolution testing run. Of primary concern is coning, which results in a cone-shaped mass of disintegrated, insoluble solids at the bottom of the Apparatus 2 vessel.
Analysis
At set time points, aliquots of filtered medium are removed and analyzed for API content by HPLC or UV-Vis. During development, HPLC is most commonly used. It has the advantage of being able to separate the API from potential interferences in the formulation matrix or dissolution medium and can detect API degradation. Furthermore, large variations in sample concentration can often be accommodated by adjusting injection volume.
Summary
Designing an appropriate dissolution method considers many API, formulation, and analytical methodology parameters. In vitro dissolution testing plays a prominent role in assuring product performance and quality. Effort should be made to investigate bio-relevant dissolution testing that mechanistically resembles in vivo conditions. Properly designed dissolution tests will accelerate drug development, hasten validation of post-approval changes, and possibly reduce unnecessary human studies.
References
- S. FDA/CDER, Guidance for Industry, “Dissolution testing of immediate release solid oral dosage forms”, 1997.
- Zhang H, Yu L, “Dissolution Testing for Solid Oral Drug Products: Theoretical Considerations”, American Pharmaceutical Review, September/October 2004, 7(5), 26-31.
- Wang Q, Fotaki N, Mao Y, “Biorelevant Dissolution: Methodology and Application in Drug Development”, Dissolution Technologies, August 2009, 6-12.
- S. FDA/CDER, Guidance for Industry: Immediate release solid oral dosage forms, “Scale-up and post approval changes: chemistry, manufacturing, and controls, in vitro dissolution testing and in vivo bioequivalence documentation”, 1995.
- USP, General Chapters <711>, <1092>, <1225>, <1088>.
- Tadey T, Carr G, “Dissolution Testing for Solid Oral Dosage Forms”, Pharmaceutical Formulation and Quality, July/August 2009, 11(4), 22-27.